
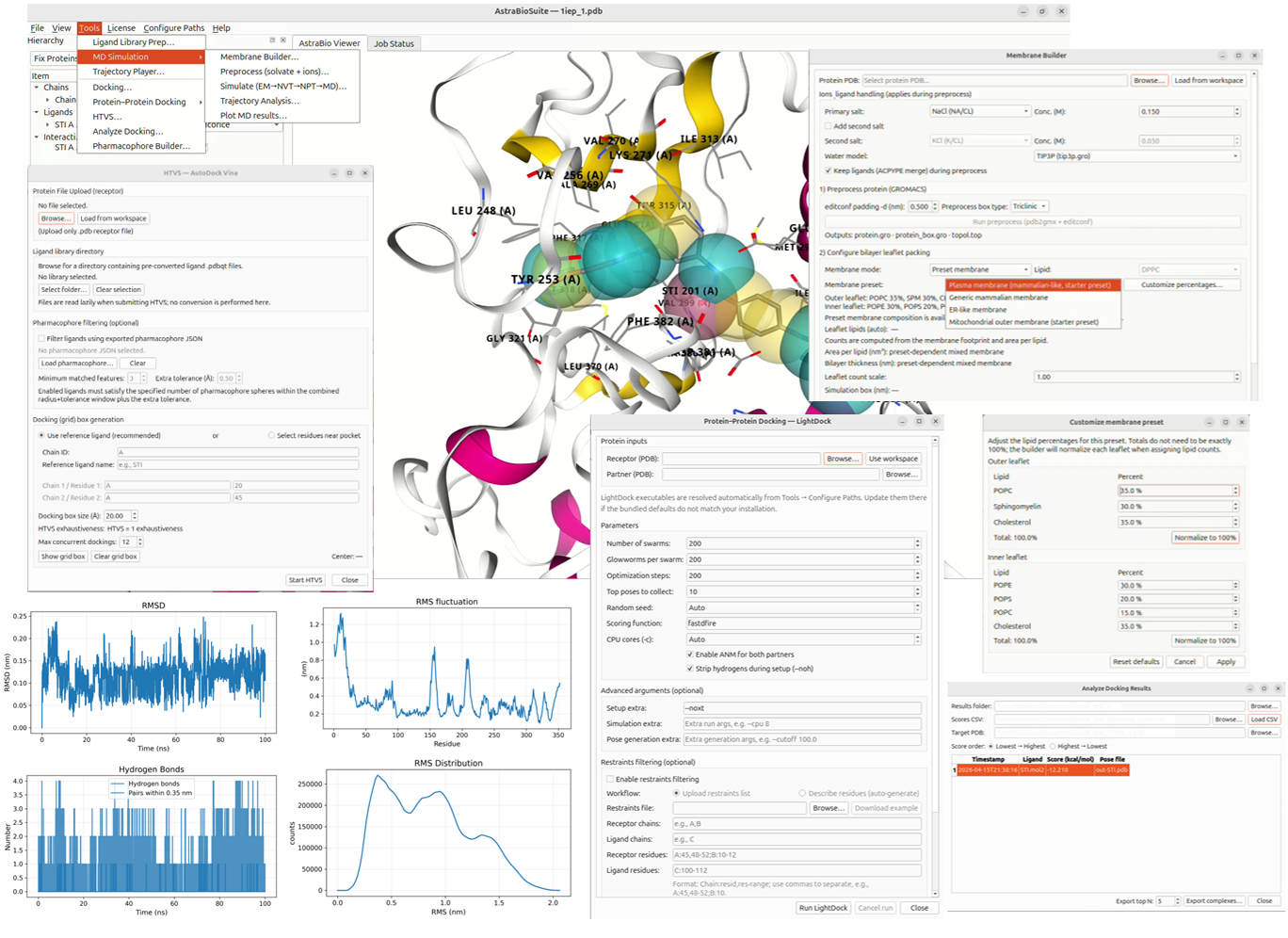
Introducing AstraBioSuite™
A Powerful and Intuitive Standalone Platform for Computational Drug Discovery
AstraBioSuite™ unifies molecular docking, HTVS, pharmacophore modeling, protein–protein docking, molecular dynamics workflow management for GROMACS-compatible simulations, trajectory analysis, and biologically relevant membrane preset workflows — including mammalian-like plasma membrane systems — into a streamlined Linux-based computational drug discovery environment.
News & Updates
Aug 16, 2025
Not every project needs a subscription. For researchers with just one or two simulations in mind, SiBioLEAD now offers cost-effective, one-time molecular dynamics plans —no ongoing fees. Click here... for more details.
Jan 20, 2023
This is to inform our valuable users that from January 20, 2023, all the SiBioLead operations and web-application will be managed and operated by the US-based firm, SiBioLead LLC. Please check Privacy Policies & Terms of Use for updated information. Changes in subscription plans and other details will be announced by the new administration.
Oct 11, 2022
Our newly designed, user-friendly MD simulation module predicts the formation of cross-β sheet structure from an alpha-helical form of Abeta42 peptide under elevated calcium level in a 300ns test simulation run.
Oct 11, 2022
Latest publication used our Diversity-based High Throughput Virtual Screening (D-HTVS) technology and identified novel dual PI3K/AKT inhibitors from 1.5 million ChemBridge compounds to control acute myeloid leukemia cell proliferations using this technology. Click here... for more reading.
Oct 07, 2023
Check out latest publication that used our GROMACS based molecular dynamic simulation platform. Click here....
About Us
Brought to you from a team of scientists and engineers.
SiBioLEAD is a premier bioinformatics Contract Research Organization (CRO) in computational drug discovery. We merge state-of-the-art technology to expedite the development of innovative therapeutics. Our interdisciplinary team, comprising computational chemists, biologists, and data scientists leverage algorithms, computational hardware, and artificial intelligence for pioneering drug discovery.
At SiBioLEAD, precision and efficiency are at the core of our mission. We Specialize in GROMACS based molecular dynamic simulations. We are committed to revolutionizing drug discovery by bridging the gap between computational modeling and experimental validation. Join us on a collaborative journey, where innovation meets impact. Together, we can translate visionary therapies into tangible realities.
Registered Users
4058Usage by Countries
89Publications
65
Our inspiring drug-discovery applications
HTVS
Our ingenious Diversity-based High Throughput Virtual Screening (D-HTVS) technology preloaded with millions of small molecules from sought-after databases, including ChEMBL, ZINC, and ChemBridge libraries, facilitates meticulous and rapid screening to successfully identify target-specific lead molecules.
Exploit our powerful cloud-servers for your rapid virtual screening.
Perform blind or unbiased docking using our SiBDock module with high precision and flexibility.
Molecular Dynamic Simulation Module
One of a kind web-application designed to set up GROMACS based macromolecular-drug bound complex simulations up to one microsecond in a single step.
Toward providing simulation service for a wide range of users, we have numerous afforable pricing plans.
Explore more ...Client-oriented services
Project-based lead-discovery services for academics and commercial entities using our powerful state-of-the art tools within stipulated time.
Our wide-range of customized services:
AI-based Protein structure modeling
Flexible Protein-protein interaction modeling
GROMACS Molecular Dynamic simulations to elucidate protein and drug dynamics
Quantitative Structure Activity Relationship (QSAR)
Virtual Kinase Profiling (kinome-wide screening) for target ligands
Our Team

Meenakshisundaram Balasubramaniam, Ph.D.
Founder & Chief Executive Officer
Phil Williams, Ph.D.
Co-Founder & Chief Technical Officer
Robert J Shmookler Reis, D.Phil.
Co-Founder & Chief Scientific Officer
Kottayil I Varughese, Ph.D.
Scientific Advisor Structural Biology & Biophysics
Prasanna Rajagopalan Ph.D., FICS.,
Senior Scientific Advisor Biological Research and BioinformaticsPricing
We offer competitive promotional pricing plans exclusive for our academic users
Free
$0 / month
- Automated ligand preparation/conformer generation
- Dock up to 2 molecules per submission
- PLIP analysis included
- No Credit Card details needed
Premium Yearly plans
- Access to our new Diversity-based High throughput virtual screening technology
- Access to pre-loaded libraries including ChemBridge, ChEMBL, NCI and ZINC compounds
- Enjoy the fully-automated, hassle-free, Molecular Dynamic Simulation module that performs simulations up to 500 nanoseconds, with extension to MMPBSA-based Gibbs binding free energy calculations.
Premium Monthly plan
- Access to our new Diversity-based High throughput virtual screening technology
- Access to pre-loaded libraries including ChemBridge, ChEMBL, NCI and ZINC compounds
- Enjoy the fully-automated, hassle-free, Molecular Dynamic Simulation module that performs simulation up to 200 nanoseconds, with extension to MMPBSA-based Gibbs binding free energy calculations
Testimonials
I have used the MD simulation module of SiBioLead, It's easy and very simple to use. Results are generated within the time. It was a great experience with SiBioLead. Thank you for developing such user friendly tool for drug discovery research.
Prasanna Sarmah
Ph.D. Scholar
CSIR-NEIST
Jorhat, Assam, India
The SiBioLead platform is an epoch-making product that greatly facilitates the experimentation of molecular dynamics simulations for beginners without programming skills. This platform will benefit many researchers and will contribute to the development of computational biology, molecular biology and new drugs.
Hong Li (Paul), BMed, PhD
Associate, School of Science
RMIT University
Melbourne, Victoria 3000 Australia
The SiBioLead interface is a ground-breaking approach that makes it substantially simpler for non-programmers to get started with molecular dynamics simulations because of its user-friendly interface and outstanding simulation runtime. As a master's student, this interface helped me finish my dissertation, which contained numerous MD simulations, in a surprisingly short period of time. They have a fantastic support system that provides the necessary files using first-rate cloud-based technologies, enabling personalized drug discovery services.
Kankipati Teja Shyam, M.Sc
Master's student
Department of Biochemistry and Molecular Biology
Pondicherry University, India
Using the molecular dynamics simulation with SiBioLead is truly handy, with easy-to-use interface and the runtime of simulation is also fast and amazing. Totally recommended to use if you have a lot of simulation to work with
Adhityo Wicaksono, Ph.D, MRSB
Postdoctoral Fellow Researcher,
Faculty of Science,
Chulalongkorn University,
Thailand
A user friendly, yet powerful tool for target docking analysis. This cutting edge technology has overcome the prerequisites of software background, while any biologist can utilize it. Thanks to SiBioleads for making an outbreaking powerful tool that support drug discovery research.
Prasanna Rajagopalan Ph.D. FICS
Chairman, Central Research Laboratory, & Assistant Professor in Biochemistry,
Department of Clinical Laboratory Sciences,
College of Applied Medical Sciences,
King Khalid University, Abha- Aseer, Kingdom of Saudi Arabia.
I used SiBDock for my project. It was effortless process. Congratulations on developing such a user-friendly tool.
Kottayil Varughese, Ph.D.
Professor,
Department of Physiology and Cell Biology,
College of Medicine,
University of Arkansas for Medical Sciences, Little Rock, AR, USA.
I have personally used SiBioLead for molecular docking. It is an awesome platform for molecular docking. It’s easy and simple to use, user-friendly and a great resource for protein-ligand docking and other computational biology projects. I recommend it for all users and the academia.
Samuel Kakraba, Ph.D.
M.S (Mathematical Sciences)., Ph.D.(Bioinformatics) Candidate and Graduate Research Assistant,
Joint Graduate Bioinformatics Program,
College of Applied Medical Sciences,
University of Arkansas for Medical Sciences & the Univ. of Arkansas at Little Rock, USA
Frequently Asked Questions
-
How to check the chain ID for my receptor PDB structure
Selecting chain IDs is crucial during the job submission process, this can be done in two ways. 1) If you are downloading your receptor (PDB) file from PDB databank, then you can check your chain ID by clicking the structure view and maneuver your mouse cursor over the structure. 2) You can view chain IDs from most of the macromolecular visulaizers including WinCoot, Avagadro, BIOVIA Discovery Studio, etc.
-
How many ligands I can submit at the same time
The current version we have (freemium) can dock up to 5 ligands at the same time. Our premium version of SiBDOCK can handle up to 100 ligands, or HTVS module can handle up to 5000 ligands (high-throughput mode) at the same time.
-
How to cite SiBiolead
If you have used our docking module, please cite the following publications in your manuscript
Trott O at al. (2010) for AutoDock Vina
Salentin S at al. (2015) for PLIP
If you have used our MD simulation module, please cite the following publications in your manuscript
Van der Spoel at al. (2005) for GROMACS
D.A. Case et al. (2015) for AmberTools22
Sousa da Silva AW et al. (2012) for ACPYPE
Valdés-Tresanco MS et al. (2021) for MMPBSA
Additionally, we request you to reference our website by providing the URL, e.g., "we used integrated docking and simulation solutions from SiBioLead (www.sibiolead.com) for all docking and simulations"; and cite one or more publications that previously cited SiBioLead.
Acknowledgements
Sibiolead modules use established open-source software





